Home > 製品情報:BIOVIA製品 > BIOVIA Discovery Studio
BIOVIA Discovery Studio
製品概要
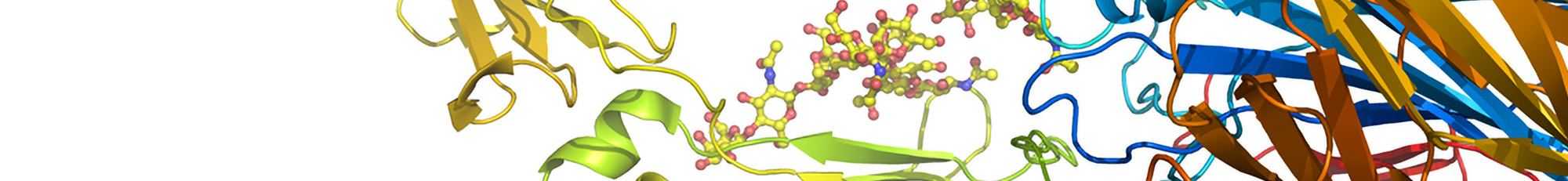
Discovery Studio は、ライフサイエンス分野で対象となるDNA、RNA、タンパク質、糖鎖、リガンド、抗体等のモデリング及びシミュレーションを行うための統合パッケージです。また、ワークフロー統合環境ソフトウェアであるBIOVIA Pipeline Pilot Serverのプラットフォーム上に構築されており、データベース検索=>シミュレーション=>結果解析=>データベース格納等の一連の処理フローを自動的に行うことが可能となっています。

主な機能
- 核酸、タンパク質等の生体高分子のモデル構築・評価、構造最適化、分子動力学シミュレーション
- 配列検索、ホモロジー検索
- ホモロジーモデリング、de novo design、ファーマコフォアモデリング
- タンパク質―リガンドドッキングシミュレーション
- 静電相互作用及び溶媒和効果の計算
- 構造活性相関計算、薬物体内動態解析、毒性予測
- 抗体モデリング
- 視覚化
適用分野
- 製薬
- 化学
- 食品
- バイオテック企業
- 大学、研究機関
製品モジュールリスト
Platform
| BIOVIA Discovery Studio Standalone | モデリング研究者向けに設計された分子モデリングプラットフォームです。モデル設計やモデリングの実行を行うのに必要な基本ソフトが組み込まれています。 |
| BIOVIA Discovery Studio Client | 入出力構造を可視化し、グラフィカルに操作を行うためのインターフェースです。BIOVIA Discovery Studioの主要なモジュールを利用するためには、BIOVIA Pipeline Pilotに接続する必要があります。 |
| BIOVIA Pipeline Pilot | Discovery Studioのモジュールを実行するためのプラットフォームです。また、プロトコルをカスタマイズすることにより独自のワークフローを構築することもできます。 |
| ActiveX Control | Windows 環境用の 3D 分子描画機能 |
Protein Modeling and Sequence Analysis
| DS SEQUENCE ANALYSIS | ホモロジー検索ソフトウェアのBLAST あるいは PSIBLAST アルゴリズムを使用した配列解析を行います。 |
| DS MODELER | 与えられた配列を既知のタンパク質立体構造を雛形にしてアラインメント及び構造最適化を行い、精度の高いタンパク質ホモロジーモデルを自動的に生成する業界標準のソフトウェアです。 |
| DS PROTEIN REFINE | CHARMm 及び自社開発のアルゴリズムを用いて、タンパク質のループ領域及び側鎖構造を最適化します。 |
| DS PROTEIN FAMILIES | 立体構造上で保持されている残基の位置とタンパク質ファミリーでの配列保持パターンを比較、解析することによって、タンパク質機能のメカニズムについて知見を得ることができます。 |
| DS PROTEIN HEALTH | Profiles-3D Verify と呼ばれる方法を用いて、モデルの構造的環境をアミノ酸の適正環境と比較することでタンパク質構造の評価を行います。 |
| DS PROTEIN DOCKING | ZDOCK アルゴリズムを使用して新しい標的のタンパク質-タンパク質構造間の相互作用を迅速かつ正確に予測することができます。 |
| DS PROTEIN AGGREGATION | タンパク質上の凝集しやすい部位の大きさと場所を特定し、安定性向上につながる変異導入箇所を予測します。 |
Biopolymer Building and Analysis
| DS BIOPOLYMER | タンパク質構造の構築と修正、構成要素の分割や整形、プロパティ解析レポートの作成、ポアソン-ボルツマン静電場 (DelPhi )で高分子または低分子の静電ポテンシャルと溶媒和エネルギーを計算するプロトコルへのアクセスを提供します。 |
Simulation
| DS CHARMM | 低分子リガンドから多成分の生体分子複合体まで、分子のエネルギーやダイナミクスを計算することができます。 |
| CFF advanced Class II Forcefield | DNA、RNA、炭水化物、脂質、タンパク質、ペプチド、低分子などのモデルを高精度で最適化します。 |
| Merck Molecular Forcefield(MMFF) | 高く評価されている Merck 分子力場を使用して、高分子とリガンド間のエネルギー特性と相互作用について研究することができます。 MMFF94 では、有機化合物や生体内低分子化合物の酵素結合に不可欠な分子間相互作用が幅広くパラメータ化されています。 |
| DS ANALYSIS | 回転運動半径 (Radius of gyration) の計算、動径分布関数やトラジェクトリのクラスタリングによる直感的で簡単な操作で、MD トラジェクトリを解析することができます。トラジェクトリのクラスタリングでは、デンドログラムのプロットや主成分解析、Phi-Psi 角の経時変化からクラスタを選択することができます。RMSD や水素結合、多数のドッキング済みリガンドに近接しているアミノ酸残基を簡単に計算することができます。 “Protein Health”ツールパネルを使用すると、タンパク質構造を確認し、異常のある領域を解析することができます。MODELER DOPE (Discrete Optimized Protein Energy) エネルギー関数に基づいて、モデルの質を評価することができます。 |
| DS QUANTUMm | 密度汎関数理論プログラムである DS DMOL3 Molecular (QM) と CHARMm (MM) を組み合わせた正確な量子力学 / 分子力学 (QM/MM) 的手法を用いることで、リード化合物の最適化におけるタンパク質-リガンドモデリングの精度を高めます。さまざまな 交換-相関汎関数や基底系を使用して、エネルギー計算や構造最適化を単一の場所で集中処理し、ドッキングアプリケーションやシミュレーションアプリケーションに正確なリガンド部分電荷を取得します。また、カチオン-π 相互作用や金属-リガンド-受容体相互作用などの特殊な相互作用を高い精度でモデル化します。簡単なジョブ設定と設定済みパラメータセットのカスタマイズによって、これらの QM/MM 法すべてにアクセスできます。 |
Receptor-Ligand Interactions
| DS FLEXIBLE DOCKING | CHARMm の正確な受容体サンプリング能力と、効率のよい特性ベースのドッキングを組み合わせた、この新しい手法を使用すると、合理的なフレキシブルドッキングを行うことができます。例えば、活性サイト内の側鎖の、既存の低エネルギーコンフォメーションの影響を受けるような低分子のドッキングを行うことができます。 |
| DS LIBDOCK | 受容体の結合サイトの極性 / 無極性 (ホットスポット) を使用すると、効率のよいドッキングを行うことができます。業界標準の Catalyst エンジンは、低分子のコンフォメーションを作成して、ドッキングの精度を高めることができます (DS Catalyst Conformation の導入をお勧めします)。 |
| DS LIGANDSCORE | 充分に検証され、洗練されたスコアリング関数とその個々の記述子で、リガンド-タンパク質相互作用を評価できます。DS LigandScore はドッキングの際の正しいポーズと誤ったポーズを区別し、予測されたリガンドに優先順位をつけることでスクリーニングや合成に役立ちます。パラメータはカスタマイズ可能で、ユーザの設定を保存することができ、他のユーザと共有できます。 |
| DS LUDI | DS Ludi は de novo 創薬アプリケーションで、新薬になりうる scaffold を迅速に同定し、受容体の結合ポケット内の相互作用部位を利用して、フラグメントライブラリからフラグメントの配置やスコアリングを実行します。ligand scaffolds ライブラリをスクリーニングしたり、受容体の結合サイト内でリガンドの置換様式を修正することができます。フラグメントライブラリは、カスタムのフラグメントを加えることも可能です。 |
| DS DE NOVO EVOLUTION | DS De Novo Evolution で、 scaffold 上のフラグメントを連結させたり構築したりすることにより、新薬になりうる分子を作成することができます。速度と精度で最適化された3つのモードから選択することができます。Quick モードでは、DS Ludi (必須) と DS LigandScore (任意) のスコアのどちらかによって順位付けが行われ、最もスコアの良い阻害剤が提示されます。Full Evolution モードでは、阻害剤の作成で最後まで残ったものが選択されます。Combinatorial モードでは、 scaffold の誘導体の組み合わせすべてがリストアップされます。 |
Pharmacophore Modeling and Analysis
| DS CATALYST BUILD & DS CATALYST SEARCH | ファーマコフォアモデルの検索に利用する 3D 化合物データベースを容易に作成できます。DS Catalyst Build を利用すると、どんな構造を持つ化合物でも、生理的条件下でエネルギー的に許容されるコンフォメーションをサンプリングして 3D データベースに保存できます。DS Catalyst Search は傑出した性能を備えており、ユーザのファーマコフォアモデル、指定された 3D 形状、部分構造を含むクエリなどを用いてデータベース検索を迅速に行うことができます。3D Catalyst データベースには詳細なコンフォメーションの情報が保存されているため、各化合物について分子の複数の幾何学的配置を検索できます。 |
| DS CATALYST HYPOTHESIS | 定性的または定量的なファーマコフォアモデルを自動的に作成し、ターゲットへの結合に必要である基本的な化学的特性や構造的特性を識別できます。ユーザは既知の一連の化合物から自動的に仮説を作り上げることが可能です。不活性分子の構造から、立体障害の存在を予測し、排除体積を規定してモデルを改良できます。また、医薬品候補や既知の活性化合物のアラインメントをもとに、生理活性を考慮せずにファーマコフォアの仮設を展開することもできます。 |
| DS CATALYST SHAPE | 分子が占めるべき形状を検索クエリーに含めることにより、受容体の活性サイトによる三次元の空間的制約を満たすことのできる化合物を見出すことができます。3D データベース検索を行い、同様の形状をもつ分子を、具体的な化学構造に関係なく特定します。全体的な形状を検討するため、検索のヒットは標準的な 2D 検索に比べるとトポロジー的多様性に富んでおり、生物学的ターゲットの形状をかなり正確に表現する一連の化合物が得られます。 |
| DS CATALYST SCORE | データベース検索から化合物を迅速に評価し、優先順位を付けることができます。データベース検索から得られたヒットの結果をユーザの仮説に当てはめ、どの化合物がユーザのファーマコフォアの要件を最も反映するかを判別します。 DS Catalyst Score は、各々の化合物について、予測フィット値あるいは活性値を算出します。ヒットリストの件数に応じて、ファーマコフォアモデルの最小限、または最大限のフィーチャ数を指定することにより、ユーザは検索結果を広げたり狭めたりすることができます。また、具体的な分子を複数並べて、分子の構造特性や化学特性を比較し、それらの分子の類似度を判別することもできます。 |
| DS CATALYST CONFORMATION | コンフォメーションモデルを迅速に算出し、これによって低分子がエネルギー的にとりやすいコンフォメーションの全てを、十分にまた多種多様に表現することができます。新しい CAESAR アルゴリズムを用いて迅速に分子が生理的条件下で取り得るリガンドのコンフォメーションを作成することができます。6つのコンフォメーションジェネレータの中から、それぞれの創薬プロジェクトに最も相応しいアルゴリズムを選べます。各アルゴリズムはユーザに合わせて最適化し、コンフォメーションのサンプルが取れるよう、パラメータをカスタマイズすることができます。 |
ADMET and Predictive Toxicology
| DS ADMET | 合成の候補やベンダーライブラリ、スクリーニングコレクションで得た分子の吸収、分布、代謝、排泄、毒性 (ADMET) の特性予測を算出することによって、化合物を早い段階で評価することができます。算出結果を用いると、合成の前に好ましくない ADMET 特性を持つ化合物を除外し、ADMET 特性向上のための分子設計に向けて構造を精密化して、評価することができます。医薬品開発の早い段階でこれらの特性を最適化することは、後の開発プロセスにおける ADMET の諸問題を軽減するために極めて重要です。DS ADMET には人間の腸内吸収、水溶解度、脳血管関門通過、血漿タンパク結合、チトクローム P450 2D6 阻害、肝毒性についてのモデルも含まれています。一連の低分子をフィルタし、 SMARTS® ルールで指定したルールに適合する分子のみを選択します。 |
| DS TOPKAT | 実験や動物モデルによって化合物の性能を評価できます。化学物質の毒性の影響や、化学物質の環境に対する影響の評価を、分子構造だけを基にして算出し評価します。TOPKAT では交差検定された、強力な定量的構造毒性相関 (QSTR) モデルを使用して、さまざまな毒性の基準を評価し、特許取得済みの Optimal Predictive Space 法(化合物が予測空間内にあることをチェックすることが可能)という検証方法を活用して結果の解釈に役立てます。 |
データシート
- BIOVIA DISCOVERY STUDIOによるタンパク質モデリングと配列解析
- AGGMAP: タンパク質凝集性向の予測
- BIOVIA DISCOVERY STUDIOでの生体高分子の構築と解析
- BIOVIA DISCOVERY STUDIO®を使用した分子シミュレーション
- BIOVIA DISCOVERY STUDIO®を使用した構造ベースの設計
- BIOVIA DISCOVERY STUDIO®を用いたファーマコフォアおよびリガンドベースの設計
- BIOVIA DISCOVERY STUDIOを用いたLIGAND-BASED DRUG DESIGN
- BIOVIA DISCOVERY STUDIO® によるQSAR、ADMET、および毒性予測
- BIOVIA DISCOVERY STUDIO®を使用した抗体開発
- BIOVIA DISCOVERY STUDIO ライフサイエンスのための包括的なモデリングおよびシミュレーション
- BIOVIA DISCOVERY STUDIOでのX線結晶解析
- BIOVIA DISCOVERY STUDIO®を使用した高分子モデリング
- BIOVIA DISCOVERY STUDIO® による高分子モデリング
- BIOVIA バイオロジクス ソリューション
- 生物製剤開発
- 化学研究開発
- ケミスト向け分子表示/解析ツール
弊社はBIOVIA製品の正規販売代理店です。
